In vivo SILAC, a comprehensive quantitative proteomics technique, evaluates the impact of PROTAC drugs on protein expression in cells, providing a closer representation of the sample's true state and reflecting real information.
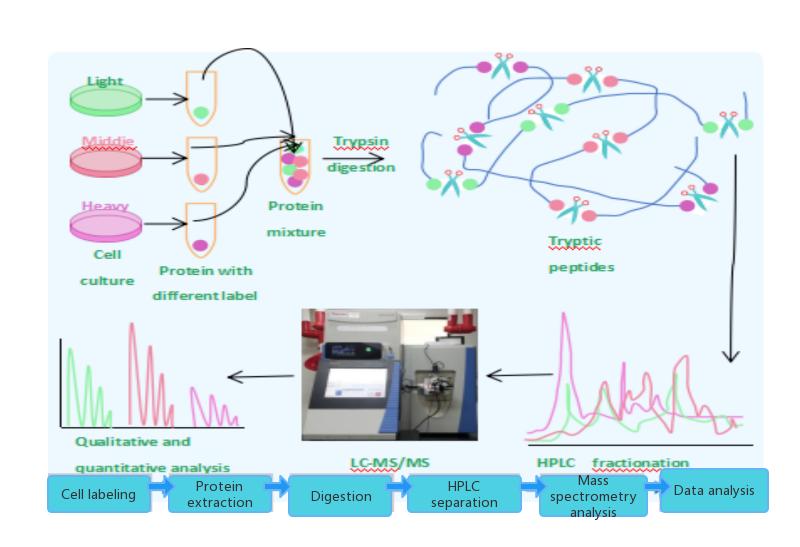
Stable Isotope Labeling by Amino acids in Cell culture (SILAC) is an effective method for studying relative proteomic changes under differential treatments. It relies on the combination of mass spectrometry and metabolic incorporation of stable isotopes into amino acids. In SILAC, a given "Light" or "Heavy" form of amino acids is introduced into the sample. Two cell populations are grown in the same medium, with one containing the "Light" form and the other containing a specific amino acid labeled with the "Heavy" form (e.g., L-lysine labeled with 12C and 13C, respectively). As the two isotopically labeled amino acids are chemically identical, their incorporation does not interfere with normal cell growth, allowing for discrimination of proteins/peptides based on their mass, making it highly suitable for mass spectrometry analysis. The SILAC method is particularly useful for monitoring post-translational modifications.
SILAC Workflow
Cell labeling Protein extraction Enzymatic digestion HPLC separation Mass spectrometry analysis Data analysis
Service Advantages
High labeling efficiency: Labeling efficiency can reach up to 100%.
High sensitivity: Requires small sample amounts, typically only a few tens of micrograms of protein per sample.
High throughput: Mass spectrometry can identify and quantify hundreds to thousands of proteins simultaneously.
High activity: In vivo labeling technology is used, providing a closer representation of the sample's true state.
High precision: Mixing multiple samples for simultaneous digestion and identification. Subsequent experiments yield consistent results, reducing the impact of experimental manipulation and equipment and ensuring higher precision and reproducibility.
Comprehensive services: Combining with other techniques, we can provide analysis of protein-protein interactions and post-translational modifications.
SILAC in PROTAC Drug Development
Changes in protein abundance induced by drugs.
Discovery of off-target proteins.
Validation of the interaction between the protein of interest (POI) and E3 ligands, among others.
Classic Case
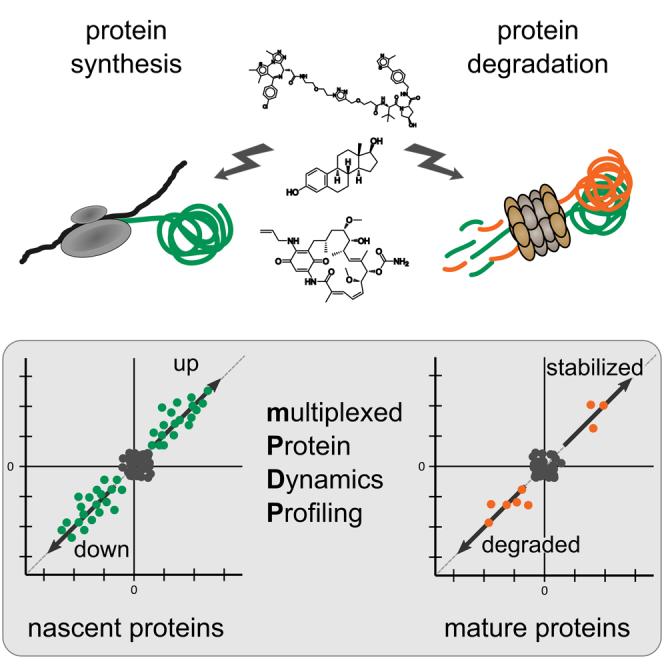
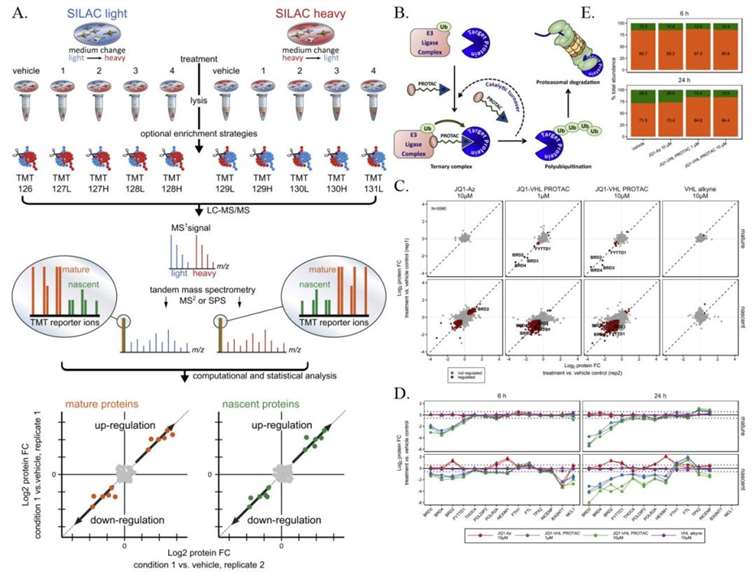 The mechanism of action of the BET degrader JQ1-VHL PROTAC
The mechanism of action of the BET degrader JQ1-VHL PROTAC
Combining SILAC with TMT chemical labeling for sensitive and comprehensive relative quantification of mature and nascent protein pools in cells. B. Schematic representation of the mechanism of action of the BET degrader JQ1-VHL PROTAC. C. Scatter plot showing protein folding changes (fold change, FC) observed in mature (top) and nascent (bottom) forms of proteins in THP-1 cells treated with BET inhibitor JQ1-Az (10μM), JQ1-VHL PROTAC (1 and 10μM), or VHL alkyne (10μM) for 6 hours, compared to the control group. D. Line graphs showing protein folding changes observed in mature (top) and nascent (bottom) forms of selected proteins in THP-1 cells treated with JQ1-Az (10μM, red), JQ1-VHL PROTAC (1μM, blue; 10μM, green), or VHL alkyne (10μM, purple) for 6 hours (left) and 24 hours (right), compared to the control group. E. Bar graph showing the relative contribution of nascent (green) and mature (orange) proteins to the protein composition based on estimated relative ion intensities after incubation with JQ1-Az (10μM) and JQ1-VHL PROTAC (1 and 10μM) for 6 hours or 24 hours.
Creative Proteomics uses proteomics methods below for PROTAC degradation study
DIA and TMT PROTACs Research
MRM/PRM PROTACs Research
AP-MS PROTACs Research
PROTACs Ubiquitin proteomics
Services You May Be Interested In
Our products and services are for research use only.

.jpg)